vcfanno

![]()
Overview
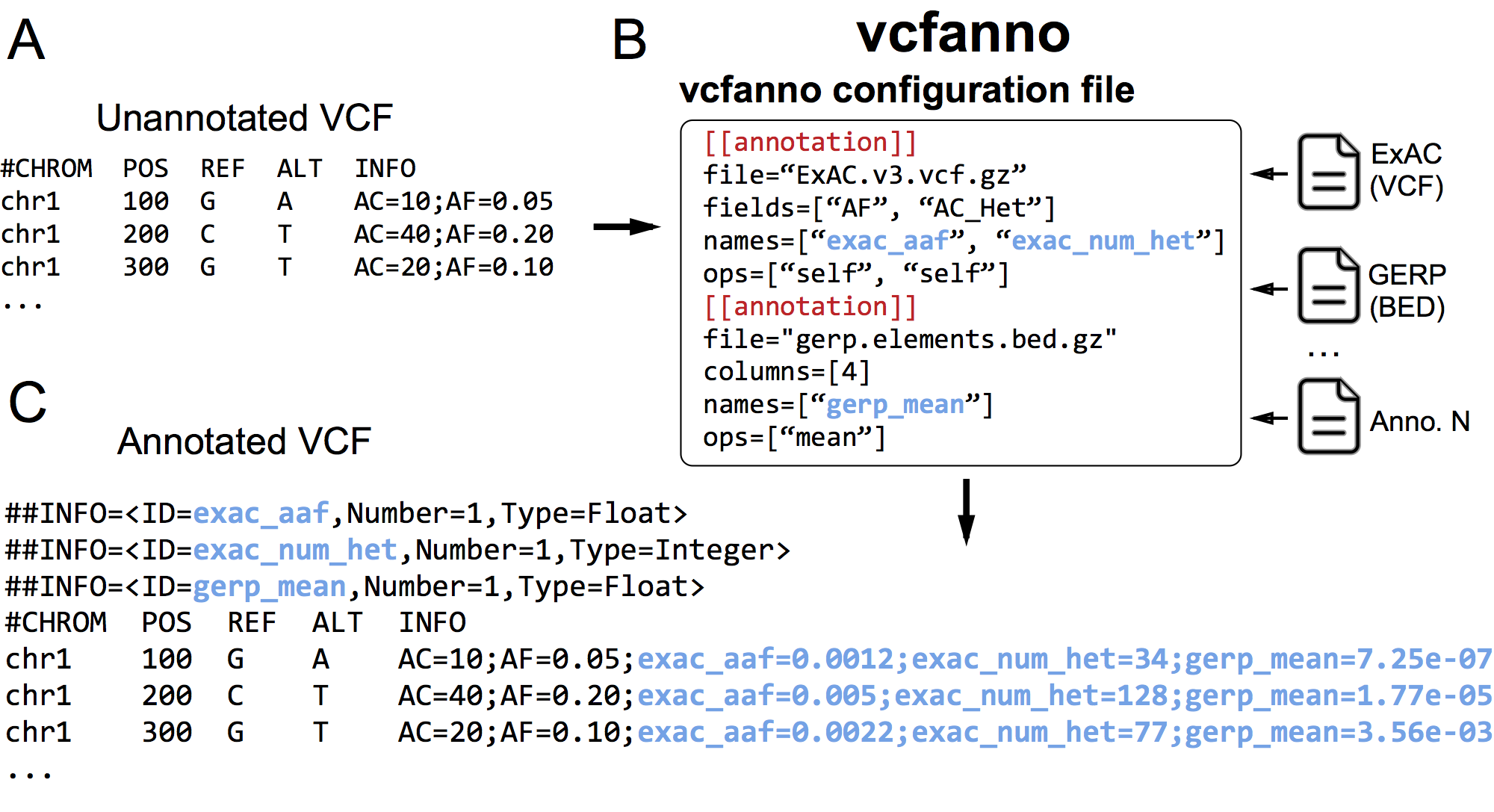
vcfanno allows you to quickly annotate your VCF with any number of INFO fields from any number of VCFs or BED files. It uses a simple conf file to allow the user to specify the source annotation files and fields and how they will added to the info of the query VCF.
- For VCF, values are pulled by name from the INFO field with special-cases of ID and FILTER to pull from those VCF columns.
- For BED, values are pulled from (1-based) column number.
- For BAM, depth (
count), "mapq" and "seq" are currently supported.
vcfanno is written in go and it supports custom user-scripts written in lua.
It can annotate more than 8,000 variants per second with 34 annotations from 9 files on a modest laptop and over 30K variants per second using 12 processes on a server.
We are actively developing vcfanno and appreciate feedback and bug reports.
Usage
After downloading the binary for your system (see section below) usage looks like:
./vcfanno -lua example/custom.lua example/conf.toml example/query.vcf.gz
Where conf.toml looks like:
[[annotation]]
file="ExAC.vcf"
# ID and FILTER are special columns is a special field that pull the ID and FILTER columns from the vcf
fields = ["AC_AFR", "AC_AMR", "AC_EAS", "ID", "FILTER"]
ops=["self", "self", "min", "self", "self"]
names=["exac_ac_afr", "exac_ac_amr", "exac_ac_eas", "exac_id", "exac_filter"]
[[annotation]]
file="fitcons.bed"
columns = [4, 4]
names=["fitcons_mean", "lua_sum"]
# note the 2nd op here is lua that has access to `vals`
ops=["mean", "lua:function sum(t) local sum = 0; for i=1,#t do sum = sum + t[i] end return sum / #t end"]
[[annotation]]
file="example/ex.bam"
names=["ex_bam_depth"]
fields=["depth", "mapq", "seq"]
ops=["count", "mean", "concat"]
So from ExAC.vcf we will pull the fields from the info field and apply the corresponding
operation from the ops array. Users can add as many [[annotation]] blocks to the
conf file as desired. Files can be local as above, or available via http/https.
See the additional usage section at the bottom for more.
Example
the example directory contains the data and conf for a full example. To run, download the appropriate binary for your system
Then, you can annotate with:
./vcfanno -p 4 -lua example/custom.lua example/conf.toml example/query.vcf.gz > annotated.vcf
An example INFO field row before annotation (pos 98683):
AB=0.282443;ABP=56.8661;AC=11;AF=0.34375;AN=32;AO=45;CIGAR=1X;TYPE=snp
and after:
AB=0.2824;ABP=56.8661;AC=11;AF=0.3438;AN=32;AO=45;CIGAR=1X;TYPE=snp;AC_AFR=0;AC_AMR=0;AC_EAS=0;fitcons_mean=0.061;lua_sum=0.061
Typecasting values
By default, using ops of mean,max,sum,div2 or min will result in type=Float,
using self will get the type from the annotation VCF and other fields will have type=String.
It's possible to add field type info to the field name. To change the field type add_intor_float` to the field name. This suffix will be parsed and removed, and your field
will be of the desired type.
Operations
In most cases, we will have a single annotation entry for each entry (variant)
in the query VCF, in which case the self op is the best choice. However, it is
possible that there will be multiple annotations from a single annotation file--in
this case, the op determines how the many values are reduced. Valid operations are:
- lua:$lua // see section below for more details
- self // pull directly from the annotation and handle multi-allelics.
- concat // comma delimited list of output
- count // count the number of overlaps
- div2 // given two values a, b return a / b.
- first // take only the first value.
- flag // presense/absence via vcf flag
- max // numbers only
- mean // numbers only
- min // numbers only
- sum // numbers only
- uniq // comma-delimited list of uniq values
- by_alt // comma-delimited by alt (Number=A), pipe-delimited (|) for multiple annos for the same alt.
There are some operations that are only for postannotation:
- delete // remove fields from the query vcf's INFO.
- setid // set the ID file of the query vcf with values from its INFO.
In nearly all cases, if you are annotating with a VCF. use self
Note that when the file is BAM, the operation is determined by the field name ('seq', 'mapq', 'DP2', 'coverage' are supported).
PostAnnotation
One of the most powerful features of vcfanno is the embedded scripting language, lua, combined with postannotation.
[[postannotation]] blocks occur after all the annotations have been applied. They are similar, but in the fields
column, they request a number of columns from the query file (including the new columns added in annotation). For example
if we have AC and AN columns indicating the alternate count and the number of chromosomes, respectively, we could create
a new allele frequency column, AF with this block:
[[postannotation]]
fields=["AC", "AN"]
op="lua:AC / AN"
name="AF"
type="Float"
where the type field is one of the types accepted in VCF format, the name is the name of the field that is created, the fields
indicate the fields (from the INFO) that will be available to the op, and the op indicates the action to perform. This can be quite
powerful. For an extensive example that demonstrates the utility of this type of approach, see
docs/examples/clinvar_exac.md.
A user can set the ID field of the VCF in a [[postannotation]] block by using name=ID. For example:
[[postannotation]]
name="ID"
fields=["other_field", "ID"]
op="lua:other_field .. ';' .. ID"
type="String"
will take the value in other_field, concatenate it with the existing ID, and set the ID to that value.
see the setid function in examples/custom.lua for a more robust method of doing this.
Additional Usage
-ends
For annotating large variants, such as CNVs or structural variants (SVs), it can be useful to
annotate the ends of the variant in addition to the region itself. To do this, specify the -ends
flag to vcfanno. e.g.:
vcfanno -ends example/conf.toml example/query.vcf.gz
In this case, the names field in the conf file contains, "fitcons_mean". The output will contain
fitcons\_mean as before along with left\_fitcons\_mean and right\_fitcons\_mean for any variants
that are longer than 1 base. The left end will be for the single-base at the lowest base of the variant
and the right end will be for the single base at the higher numbered base of the variant.
-permissive-overlap
By default, when annotating with a variant, in addition to the overlap requirement, the variants must share the same position, the same reference allele and at least one alternate allele (this is only used for variants, not for BED/BAM annotations). If this flag is specified, only overlap testing is used and shared REF/ALT are not required.
-p
Set to the number of processes that vcfanno can use during annotation. vcfanno parallelizes well
up to 15 or so cores.
-lua
custom in ops (lua). For use when the built-in ops don't supply the needed reduction.
we embed the lua engine go-lua so that it's possible to create a custom op if it is not provided. For example if the users wants to
"lua:function sum(t) local sum = 0; for i=1,#t do sum = sum + t[i] end return sum end"
where the last value (in this case sum) is returned as the annotation value. It is encouraged
to instead define lua functions in separate .lua file and point to it when calling
vcfanno using the -lua flag. So, in an external file, "some.lua", instead put:
function sum(t)
local sum = 0
for i=1,#t do
sum = sum + t[i]
end
return sum
end
And then the above custom op would be: "lua:sum(vals)". (note that there's a sum op provided
by vcfanno which will be faster).
The variables vals, chrom, start, stop, ref, alt from the currently
variant will all be available in the lua code. alt will be a table with length
equal to the number of alternate alleles. Example usage could be:
op="lua:ref .. '/' .. alt[1]"
See example/conf.toml and example/custom.lua for more examples.
Mailing List
Installation
please download a static binary (executable) from here and copy it into your '$PATH'. There are no dependencies.
If you use bioconda, you can install with: conda install -c bioconda vcfanno
Multi-Allelics
A multi-allelic variant is simply a site where there are multiple, non-reference alleles seen in the population. These will
appear as e.g. REF="A", ALT="G,C". As of version 0.2, vcfanno will handle these fully with op="self" when the Number from
the VCF header is A (Number=A)
For example this table lists Alt columns query and annotation (assuming the REFs and position match) along with the values from the annotation and shows how the query INFO will be filled:
| query | anno | anno vals | | | ALTS | ALTS | from INFO | result | | ------ | ---- | ---------- | ------- | | C,G | C,G | 22,23 | 22,23 | | C,G | C,T | 22,23 | 22,. | | C,G | T,G | 22,23 | .,23 | | G,C | C,G | 22,23 | 23,22 | | C,G | C | YYY | YYY,. | | G,C,T | C | YYY | .,YYY,. | | C,T | G | YYY | .,. | | T,C | C,T | AA,BB | BB,AA | # note values are flipped
So values that are not present in the annotation are filled with '.' as a place-holder.